熱線:021-56056830,66110819
手機:13564362870
熱線:021-56056830,66110819
手機:13564362870
2結果與討論
2.1“干燥”[BMIm]BF4體系
已有研究表明,PMBA在SPR催化下發生脫羧生成TP分子,SPR是驅動催化反應的決定性因素.且該反應為二級反應,根據二級反應動力學公式:
式中:[I996cm-1]t表示反應到t時刻時,歸屬于TP的996 cm-1處環面內彎曲振動峰強度;k(s-1)為該反應的表觀速率常數,與二級反應速率常數k2(L·mol-1·s-1)成正比例關系;a為比例系數.以歸屬于產物TP的996 cm-1處環面內彎曲振動峰與歸屬于反應物PMBA的1585 cm-1處C—C伸縮振動峰的相對強度(I996cm-1/I1585cm-1)隨時間變化的曲線進行擬合獲得反應速率常數.PMBA脫羧反應明顯受電極電位的影響,在激光照射下,在較負的電位區間通常不發生反應,在相對較正的電位區間內可發生反應.因此,實驗可采用電位階躍法研究電極電位對PMBA脫羧反應的影響,即,從不發生反應的電位階躍至不同的反應電位,并連續測試SERS光譜,記錄反應變化過程。

在“干燥”[BMIm]BF4體系(經干燥預處理,無額外水)中,在-1.0 V(vs.Pt,除特別說明外,以下均相同)下保持100 s后,立即將電位階躍至-0.3 V,得到PMBA隨時間變化的SERS譜圖[圖1(A)和(B)以及TP與PMBA譜峰的相對強度隨時間變化的曲線[圖1(C).值得說明的是,該電極的SERS活性和均勻性均符合數據的橫向和縱向對比的要求,便于在考察單變量因素下的界面脫羧反應(電極的詳細性能見本文支持信息圖S1)。
在圖1(A)和(B)中,在-1.0 V保持的100 s區間內,并未觀察到位于996 cm-1處歸屬于產物TP的特征峰,說明此時PMBA未發生SPR催化脫羧反應;當電位階躍至-0.3 V后,996 cm-1處開始出現新峰,說明此時脫羧反應發生.為了測定該實驗條件下的反應速率常數,對產物與反應物譜峰的I996cm-1/I1585cm-1隨時間變化的曲線進行擬合[圖1(C).可見,在-1.0 V保持100 s時間內,I996cm-1/I1585cm-1的值幾乎不發生變化,此時反應速率常數為零,即未發生脫羧反應;當電位由-1.0 V階躍至-0.3 V時,I996cm-1/I1585cm-1值隨時間的延長逐漸增大,500 s時增大至0.05,通過二級反應動力學公式擬合分析,得到該條件下的反應速率常數約為0.12 ms-1。
將電位由-1.0 V分別階躍至-0.8,-0.6,-0.5,-0.4,-0.3和-0.2 V,結果如圖S2所示。

可見,電位從-1.0 V階躍至-0.8 V時,脫羧反應緩慢發生,反應速率常數為0.015 ms-1,隨著電位繼續向-0.6,-0.5,-0.4,-0.3及-0.2 V階躍,反應速率常數依次為0.018,0.090,0.094,0.120和0.340 ms-1,由此說明隨著階躍電位正移,反應速率常數逐漸增大。
為了進一步研究反應速率常數與電極電位的關系,分別以反應速率常數k和反應速率常數的對數lnk對階躍電位作圖(圖2).可見,k與所施加的電極電位成指數關系[圖2(A),lnk與所施加的電極電位呈現較好的線性關系[圖2(B).從電化學-SERS體系的角度,電位可以改變電極界面反應的活化能(ΔG),即ΔG=-ZEF(其中,Z為轉移的電子數;E為施加的電極電位;F為法拉第常數).從熱力學角度看,ΔG=-RTlnK(其中,K為反應平衡常數;T為溫度;R為氣體摩爾常數).將上述兩個公式結合起來,得到lnK=ZFE/RT,即lnK與E呈線性關系,且這一結論在水溶液體系與離子液體體系均成立,這也說明該反應的速率常數與平衡常數之間存在一定的定量關系,但需要全面解析反應機理后才能得到相應的定量關系。
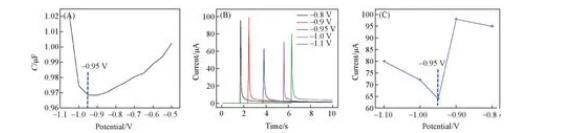
為了進一步測定PMBA在離子液體中發生催化脫羧反應的電極電位,對[BMIm]BF4的pzc進行測試.分別采用微分電容法6和計時電流法(i-t曲線法)進行測定,具體實驗方法見本文支持信息.圖3(A)~(C)分別給出了使用微分電容法和i-t曲線法測得的“干燥”[BMIm]BF4/Au MLF GC的零電荷電位。
根據Lockett等7和Ohsaka等8的相關報道以及Baldelli9和Nanbu等0的原始光譜數據,微分電容曲線的局部最低點對應的電極電位可被認為是零電荷電位,據此“干燥”[BMIm]BF4/Au MLF GC體系的pzc約為-0.95 V[圖3(A).值得說明的是,出現瞬時電流的時間僅與電極浸入的時間點有關,與預控電位無關[圖3(B).圖3(C)為瞬時電流與預控電位的關系,可見,在-0.95 V時的極化電流最小.綜合兩種測試方法,可認為Au MLF GC電極在“干燥”[BMIm]BF4體系的pzc在-0.95 V左右。
在“干燥”[BMIm]BF4中,PMBA在-1.0 V時未發生脫羧反應,而在-0.8 V時有產物TP生成,由此推測,PMBA在pzc以負的電位區間不發生脫羧反應,通常pzc以負電位區間,咪唑陽離子吸附在電極表面,空間位阻效應較大,水分子難以到達電極界面參與反應;而在pzc以正的電位區間,體積較小的陰離子吸附在電極表面,而體系中極微量水分子更傾向于與陰離子結合接近電極表面,導致了PMBA的催化脫羧反應發生,以上結果說明,離子液體陰陽離子種類和結構對反應也將產生影響。
由此也可推測PMBA在含痕量水離子液體體系中的脫羧反應機理.在pzc以正的電位作用下,電極表面呈現正電荷,離子液體陰離子吸附在電極表面,且體系中微量水分子受氫鍵作用靠近電極表面.與此同時,在激光作用下,Au MLF發生表面等離子體共振,產生SPR效應,PMBA中的—COOH脫質子轉化為—COO-,靠近電極表面,激發的熱空穴將—COO-活化形成芳基自由基,而熱電子奪取水中的質子形成氫自由基,最后二者結合生成產物TP。
2.2含水量對脫羧反應的影響
在[BMIm]BF4中加入不同摩爾分數(XW)的水,研究了PMBA脫羧反應與所施加電位階躍的關系.圖S3和圖S4(見本文支持信息)分別為[BMIm]BF4中外加XW=0.001和XW=0.003的水時,采用電位階躍法,I996cm-1/I1585cm-1隨時間變化的曲線.為了更直觀地進行對比,比較了PMBA在“干燥”[BMIm]BF4體系和不同含水量[BMIm]BF4體系中不同階躍電位后的反應速率常數,結果列于表1。

由圖S3和圖S4可以看到,由于外加水的存在,體系的pzc負移,即更易發生脫羧反應,因此選擇的起始階躍電位負移.當施加的電極電位過正時,在電位階躍瞬間,反應迅速發生后出現歸屬于產物TP的996 cm-1的譜峰強度出現下降趨勢,其主要原因來自于TP在較正電位下的脫附以及基底電極的SERS效應下降,此時不再向更正電位方向階躍。

由表1的縱向對比可見,在同一階躍電位下,隨著離子液體體系含水量增加,反應速率常數明顯增大,這與體系中界面水的增加有關,也證明PMBA的脫羧反應強烈依賴于水的存在;同時,隨體系含水量增加,脫羧反應發生的電位區間變窄,這是因為離子液體的電化學窗口隨體系含水量的增加而變窄.圖4(A)和(B)分別給出了[BMIm]BF4體系外加XW=0.001和XW=0.003水時,在不同階躍電位下,脫羧反應達到平衡時PMBA脫羧反應的情況,對比了不同階躍電位下反應達到平衡時(即TP信號最強時)的SERS譜圖。
由圖4(A)可以看到,當外加水XW=0.001,電位階躍至-0.8和-0.7 V時,反應在200 s后達到平衡,但此時996 cm-1處歸屬于產物TP的譜峰的相對強度仍然較弱,說明脫羧反應的效率較低.隨著電位向正電位方向階躍,反應達到平衡所需時間由200 s縮短至100 s,且996 cm-1處譜峰的相對強度更加明顯,說明隨著階躍電位的正移,脫羧反應更容易發生且反應進行的程度更大。
由圖4(B)可以看到,當體系外加水XW=0.003時,隨著階躍電位正移至-0.4 V,反應達到平衡所需時間由200 s顯著縮短至80 s,說明隨著體系外加水含量的增加,脫羧反應得以更快發生.由此可見,PMBA的脫羧反應的速率和效率與水密切相關,其中主要是水中的質子源參與了脫羧反應后加氫生成TP的反應。
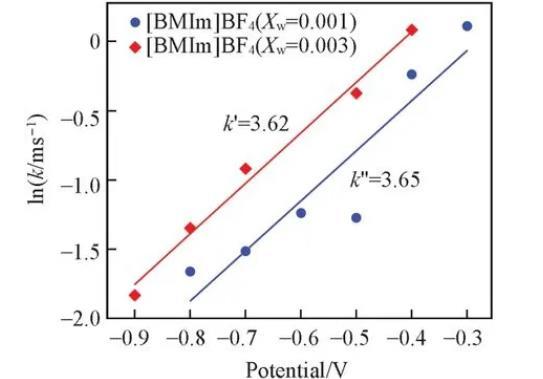
圖5比較了XW=0.001和XW=0.003體系中lnk與階躍后電位的斜率.可以看到,在[BMIm]BF4(XW=0.001)體系中,lnk與階躍后電位的斜率k′為3.62,在[BMIm]BF4(XW=0.003)體系中,lnk與階躍后電位的斜率k′′為3.65,二者幾乎相同,證明體系外加不同含量水時,lnk與階躍后電位均呈現相同的線性關系。
3、結論
采用電位階躍法,以Au MLF GC電極作為SERS基底,研究了PMBA在不同含水量的[BMIm]BF4體系的反應動力學行為.結果表明,干燥的[BMIm]BF4的pzc為-0.95 V,PMBA在離子液體體系中脫羧反應的發生與體系的pzc密切相關,在pzc以負電位區間幾乎不發生反應,而pzc以正區間脫羧反應順利發生;隨著體系水含量增大,反應速率常數增大,且外加不同含水量時,反應速率常數的對數與所施加的電極電位均呈相同的線性關系.研究結果為離子液體/金屬電極界面反應過程的動力學研究提供了詳細的信息,同時,電化學SERS光譜結合有序金納米粒子單層膜電極,可發展成為電化學界面研究的可靠的高靈敏度工具之一。
